精读笔记
Problem Setting
[Resolving superposition in AI for interpretability and cross-modal alignment in patient-neuronal images](arXiv preprint / 2026)
这篇论文实际处理的是 biological image representation 的“度量可信度”问题,而不是单纯解释 CNN 分类。作者认为在高维、稀疏、连续的生物状态空间中,神经网络为了在低维 latent 中容纳大量概念,会把多个概念压到非正交方向;这会导致两个后果:feature map polysemantic,解释不稳定;更重要的是 latent space 的距离、邻域、轨迹和跨模态对齐都会被污染。
以前的 Grad-CAM/SHAP 类方法卡在 attribution non-uniqueness:在 manifold 上做分量归因没有唯一答案,不考虑 manifold 又会产生 off-manifold explanation。论文的关键矛盾是:下游生物学分析高度依赖 representation geometry,但原始 CNN latent geometry 可能正是最不可信的部分。
Motivation
作者的动机不是“SAE 可解释,所以用 SAE 解释图像”,而是更强的判断:如果 superposition 改变了 latent metric,那么所有基于距离的生物图像分析都可能在错误几何上运行。这个缺口在生物图像里尤其明显,因为真实 biological state 数远大于模型 hidden dimension,而且 marker、organelle morphology、cell death、mutation effect 之间强相关,正是 superposition 最容易发生的场景。
论文真正缺的是一个 intrinsic alternative:不再问“某个输出由哪些像素贡献”,而是先把模型内部压缩表征重写成一个稀疏、可分解、可度量的状态空间。这个动机比常规 XAI 更接近 representation learning / manifold learning 的问题定义。
Core Idea
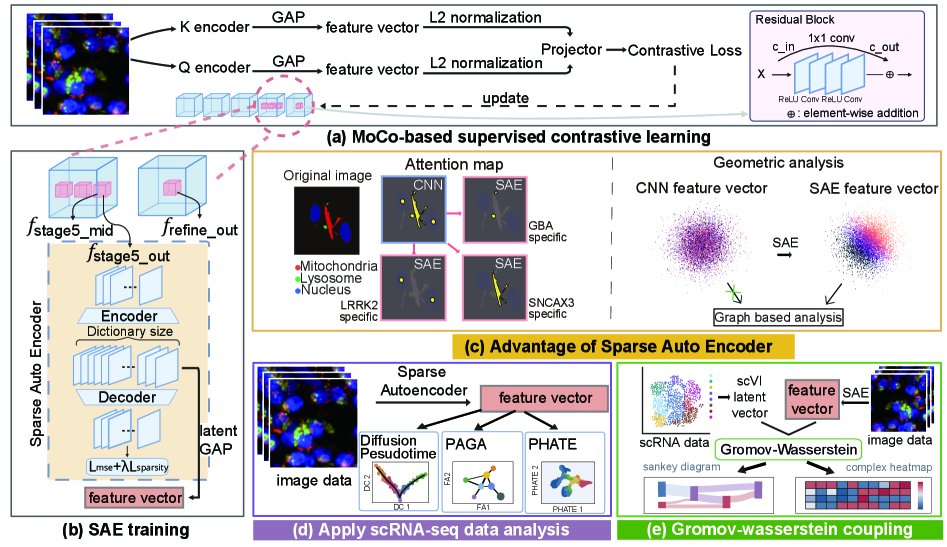
核心思想是:把 CNN 的局部 feature tokens 视为被压缩的 concept mixture,用 overcomplete gated SAE 学一个高维稀疏字典,把原本重叠在同一方向上的生物概念拆成多个更接近单义的 latent coordinates。这样引入的 inductive bias 是“高维白空间 + 稀疏激活”:概念不必共享同一 dense axis,而是可以以低 overlap 的方式分布在更大的字典中。
这和 prior 的本质区别在于,它不是在原 latent 上做解释,也不是训练一个显式 concept bottleneck,而是把已有图像表征重新参数化为一个更适合几何操作的稀疏状态空间。随后作者把这个空间当作 pseudo-transcriptomic profile,迁移 scRNA-seq 的 graph/trajectory 工具,并用 GW OT 依赖结构相似性而非 paired labels 做 image-RNA coupling。可扩展性来自一个假设:只要图像和转录组是同一 biological state 的不同投影,其内部几何在去混叠后应当可对齐。
Method
方法上真正必要的机制只有几个。第一,contrastive CNN 提供一个足够强的 morphology encoder;它不是主要创新,但确保 latent 中已有 mutation 和 cell death 相关信息,否则 SAE 只能稀疏化噪声。第二,选择中间层而非最终 refine layer,是为了避开 terminal collapse;最终分类友好的 latent 不一定是生物状态友好的 latent。第三,gated SAE 用高维 sparse dictionary 解开局部 token 的 polysemanticity,核心变化是把 dense channel space 转成 sparse concept activation space。
第四,L2 normalization / instance normalization 主要是在压制 cell density 等显微成像 confounder;这是必要 engineering,但不应和 superposition resolution 混为一谈。第五,PHATE/DPT/PAGA 不是新算法,它们在这里的作用是 stress test:如果 SAE 表征真有单细胞状态空间性质,那么这些依赖邻域图的工具应能产生连续且有生物含义的轨迹。第六,GW-map 用 Gromov-Wasserstein 对齐 image SAE space 和 scVI space,关键是只匹配 intra-modal geometry,因此不需要 paired spatial transcriptomics。
Key Insight / Why It Works
最重要的 insight 是:superposition 的代价不只是“feature 不可解释”,而是“metric space 不可用”。如果多个概念共享非正交方向,那么 cosine/L2/kNN 中的相似性会混入 concept collision 和 repulsion;下游 manifold analysis 看到的邻域不再是 biological neighborhood,而是模型压缩策略的副产物。SAE 有效的原因不是神秘的 interpretability,而是它用 overcomplete sparse basis 增加表示自由度,降低 activation overlap,让距离更接近由少数 active concepts 决定。
我认为论文最核心的贡献是把 mechanistic interpretability 的 superposition 语言转成 biological representation geometry 的问题,并用单细胞分析工具作为几何恢复后的下游验证。这比“SAE feature map 更好看”更重要。相对而言,GW-map 是有价值的应用延伸,但其增益来源更复杂:可能来自 SAE 几何改善,也可能来自 genotype structure、scVI latent smoothing、PCA 降噪、GW 的结构匹配能力以及后续非线性 regressor 的 capacity。这里增益来源不清。
哪些可能只是 engineering / scaling:更大的 SAE dictionary、PCA/standardization、L2 norm correction、batch-balanced contrastive training、scVI denoising、MLP/XGBoost gene prediction都可能贡献显著收益。论文把这些串成一个强 pipeline,但并没有完全隔离每个因素。尤其是“SAE 恢复真实几何”这个 claim,目前更多是由 proxy 指标和 biological plausibility 支撑,而不是由 ground-truth geometry 直接验证。
Relation To Prior Work
这篇工作处在三条谱系的交叉点:mechanistic interpretability 的 SAE / monosemanticity,cell morphology representation learning,以及 single-cell multi-omics alignment / GW OT。和 Grad-CAM/SHAP 的差异不是解释形式不同,而是解释对象不同:它不解释输出决策,而是试图重构一个可解释且可度量的 latent state space。
和已有 SAE 工作相比,实质新增是把 superposition 的危害从 interpretability 推到 metric contamination,并在真实生物图像上验证。和 CellProfiler/weakly supervised morphology embeddings 相比,它不只是学习表型 embedding,而是声称 embedding 需要先经过 sparse geometric purification 才能用于单细胞式分析。和 spatial transcriptomics prediction / histology-to-gene 方法相比,它没有 paired spatial RNA supervision,而是用 GW 结构对齐;这点是实质创新,但也更依赖两侧 manifold 真共享结构这一强假设。
看似新的部分中,PHATE/DPT/PAGA/GW/scVI/SAE 都是已有组件,论文的新意主要在组合逻辑和问题重定义:把 SAE 作为跨模态 biological state alignment 的前置几何校正器,而不是单纯解释工具。
Dataset / Evaluation
数据是 patient-derived iPSC cortical neurons,高内涵活细胞成像,包含健康与三类 PD mutation,规模超过十万 patch;这比 toy interpretability 设置更接近真实生物图像问题。evaluation 覆盖了分类、未监督 cell death proxy、局部邻域一致性、轨迹分析、跨模态 scRNA 对齐和 pathway plausibility,整体上能支持“这个表征对生物分析更有用”。
但它没有真正完成跨场景验证:主要还是同一实验体系、同一疾病方向、同一成像 protocol 内部闭环。GW-map 使用的 scRNA-seq 来自 SNCA 3D organoid,图像来自 2D culture,作者过滤为 2D-equivalent populations,但这既是必要处理,也可能引入选择偏差。核心 claim“superposition contaminates metric geometry”缺少独立 ground-truth manifold;cell death、Moran’s I、DPT 相关性都是合理 proxy,但不是决定性证据。
跨模态部分的评估尤其需要谨慎。基因 predictability 和 pathway clustering 有生物合理性,但这类 plausibility 容易被后验解释放大;没有 perturbation、spatial transcriptomics 或 held-out biological intervention 时,很难判断 GW-map 是否真的恢复了 causal molecular state,还是捕捉了 genotype-conditioned morphology-transcriptome correlation。
Limitation
方法成立依赖几个强前提。第一,CNN latent 中必须已经编码了足够丰富的生物状态;SAE 不能凭空发现未编码信息。第二,SAE 学到的 sparse axes 必须接近稳定 biological concepts,而不是 dataset-specific visual primitives;文中通过 AM 和 feature maps 做了支持,但文中未充分说明跨 seed、跨 batch、跨实验体系的 feature identity 稳定性。第三,图像 morphology 和 transcriptome 必须共享可对齐的 intrinsic geometry;这是 GW-map 的核心假设,也是最脆弱的假设。
scalability 上,overcomplete SAE 和 GW OT 都不便宜;如果扩展到多模态、多病种、百万级单细胞和更高分辨率图像,计算和内存会成为实际瓶颈。泛化也尚未真实证明:作者承认 ViT、其他 modality、其他 clinical domain 未验证。所谓“几何恢复”可能部分来自维度扩张和稀疏正则的通用降噪效应,而不一定是严格意义上的 superposition disentanglement。
最关键的限制是问题可能被转移了:原来是在 CNN latent 中解释混叠概念,现在变成解释 SAE 字典、选择 sparsity threshold、决定哪些 feature 是 biological concept。这个转移有价值,但不是完全消除非唯一性;dictionary learning 本身仍可能存在旋转、重构等价和超参数依赖。
Takeaway
- 第一,值得记住的不是“SAE 可以解释神经元图像”,而是“如果 latent geometry 被 superposition 污染,所有基于邻域/距离的生物表征分析都应先被怀疑”。
- 这对 cellular imaging、pathology foundation model、morphology embedding 都有迁移价值。
- 第二,SAE 在这里更像 representation geometry repair layer,而不是 visualization tool。
- 未来有价值的方向是把 sparse expanded representation 作为标准中间层,用于 clustering、trajectory、retrieval、cross-modal alignment,而非只生成可视化热图。
一句话总结
这篇论文把 SAE 从“解释模型内部 feature”的工具推进为“修复生物图像 latent geometry 并支撑无配对跨模态对齐”的前置表示层,真正贡献在于把 superposition 问题从可解释性扩展到 representation metric fidelity。